
NEW FRONTIER: “THE PLACE OF GENETICS IN SICKLE CELL DISORDER”

A Tribute To The Late DR. (MRS.) GRACE ENANG UKPONG
June 16, 2023
DONATION OF ESSENTIAL ITEMS FOR SCHOOLS’ QUIZ BY LA ROCHE LEADERSHIP FOUNDATION
June 16, 2023
NEW FRONTIER: “THE PLACE OF GENETICS IN SICKLE CELL DISORDER”
By Dr Ann Abiola Ogbenna
Sickle cell disease (SCD) is a group of inherited red blood cell disorders. It results from the inheritance of at least one copy of sickle haemoglobin (HbS) and a second copy of a gene encoding HbS or another abnormal haemoglobin. It is one of the most common severe monogenic disorders in the world. Globally about 275,000 infants are born annually with SCD. Of these, 200,000 are born in Africa.1
To understand the physiologic impact of the genetic change in SCD, we must understand how healthy red blood cells work. Healthy red cells are round and flexible allowing them to move throughout the body to deliver oxygen. They contain a protein called haemoglobin which carries the oxygen. There are 2 major types of haemoglobin, the adult haemoglobin (HbA) and the foetal haemoglobin (HbF). Each haemoglobin molecule has four globin chains with an iron molecule in the centre of each haemoglobin. The various forms of haemoglobins are named according to the types of globin chains present. (Table 1) HbF holds onto oxygen better than HbA, so we say it has a higher affinity for Oxygen (O2). The genetic mutation in SCD affects the beta chain of the adult haemoglobin, called HbS.
| Table 1: Types of healthy Haemoglobins | ||
| Globin Chains | Type of Haemoglobin | Function |
| alpha2, beta2 | Haemoglobin A | Adult |
| alpha2, delta2 | Haemoglobin A2 | Adult |
| alpha2, gamma2 | Haemoglobin F | Foetal-Neonatal |
The gene to produce alpha globin chain is located on chromosome 16 while that of the beta, gamma and delta chains are located on chromosome 11.
HbS is caused by a point mutation in the β-globin gene in which the 17th nucleotide is changed from thymine to adenine. This single mutation changes the sixth amino acid in the β-globin chain from glutamic acid to valine.2 The amino acid change makes Hb to become insoluble when oxygen is low, and they stack on each other (polymerise) in the RBC making the cell red blood cells to become stiff and sticky, looking like a C-shaped farm tool called a “sickle.”3 The stiff and sticky RBC cannot cross tiny vessels. This leads to lack of oxygen to the tissue. It is this lack of oxygen, called hypoxia that causes pain (vaso-occlussive crisis) and the short half-life of the RBCs.
At birth till about the sixth month of life, the predominant Hb is the HBF. Symptoms of SCD start at about the sixth month of life when the production of gamma globulin chain is replaced by the faulty beta globulin chain. HbF has an anti-sickling effect and can reduce or possibly eliminate haemoglobin polymerization if in sufficient quantity within the erythrocyte.
The landscape of SCD treatment has continued to evolve rapidly. Currently, there are several disease-modifying therapies in development, but cure can only be proffered if the faulty gene can be replaced, repaired, or changed. Genetic therapies can modify faulty genes and offer the possibility of cure.
Changing the gene in SCD
There are two broad ways of changing the faulty sickle gene. Both involve haemopoietic stem cells (HSC). HSC are precursor cells which can turn into any blood cell in the body.
The first way is by replacing the faulty gene with a healthy gene obtained from somebody (called the donor) else’s HSCs. The bone marrow contains haematopoietic stem cells. The HSCs contain the genes which determine the type of red blood cell produced by the body. This means if the faulty HSC producing sickling haemoglobin (Hb) is removed and replaced with a normal HSC from a donor, SCD can theoretically be cured. The donor HSCs are used as vehicles to deliver the healthy Hb gene. This is what is done in haemopoietic stem cell transplantation.
However, there are challenges to applying this procedure to every patient with SCD. First, is the fact that a compatible donor must be found. Less than one third of patients with SCD can find a suitable donor.4 Secondly, despite the compatibility, there are still high risks of complications which include, graft failure, graft-versus -host disease, delayed immune reconstitution, infertility, secondary tumours. Despite improvements in this procedure, the risk of GVHD cannot be completely removed.5
The need for compatible donors and the risk of immunogenic complications can hence be avoided if “good” autologous (self) Hb genes are given back to a patient rather than an “allo” (donor) gene.
The faulty gene can also be changed or repaired through gene therapy. Gene therapy is the introduction, removal or change in genetic material specifically DNA or RNA in the cell of a patient to repair or change a gene to treat a specific disease.6 In SCD, it addresses the genetic cause, which is the faulty gene. It uses a person’s own genetically modified HSC to produce more foetal Hb instead HbS or to produce healthy adult Hb.
The first step in gene therapy is the stem cell collection, followed by genetic modification of the harvested cells. Subsequently, the patient receives chemotherapy to eliminate the HSCs carrying the faulty gene. This makes room for the genetically manipulated stem cells which will be injected back to the patient. At the end, healthy self-red blood cells are produced
The harvested stem cells are used for genetic manipulation of the faulty gene and as backup of patients HSC. The different types of gene therapy have different genetic target sites. (Table 1) One of the most common target sites is the BCL11A gene.
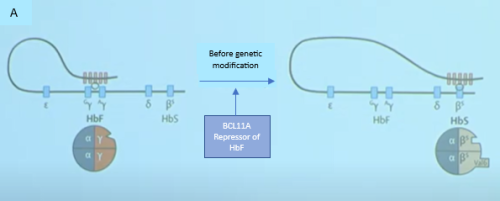
BCL11A is a major transcription factor which regulates the switch from gamma globin to beta globin after birth.7 It represses the production of gamma globin and hence HbF while allowing the production of beta globin and hence Adult Hb. In a patent with SCD, If the BCL11A is inactivated, the repression of the gamma globin will be removed, and the patient can once again start to produce HbF. HbF prevents red cells from sickling. See Figure I.
 |
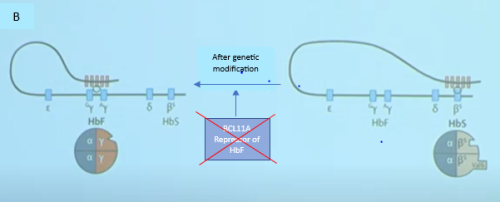 |
| Figure 1: Effect of gene modification on fetal haemoglobin switch.10
In figure 1a an active BCL11A gene suprresses ϒ-chain production, hence HbF production while favouring the production of β-globin which carries the mutated gene (HbS). In Figure 1B, the BCL11A gene has been edited or siilenced and there is switch back to the production of fetal Hb |
There are four types of gene therapy
- Gene editing: This type of genetic therapy uses a precise cut in the DNA to ‘correct’ the faulty gene directly inside the cell. Examples are therapies targeting the BCL11Agene, a negative regulator of HbF. Gene editing is used to turn off the regulation of HbF in order to increase HbF production.
- Gene silencing: This form of gene therapy supresses the expression and hence production of specific gene products. It has also been used to suppress the expression of BCL11A gene hence increasing the production of HbF. Unlike gene editing, this modification is done ex-vivo and hence needs a viral vector for delivery.
- Gene addition: Here the native HbS gene is not altered rather a new genetically non-sickling Hb gene is delivered into the stem cells using a viral vector.8
- Gene correction: This form of gene therapy is the only type that currently aims to eliminate HbS production and introduce a non-sickling haemoglobin simultaneously.
Clinical trials are currently ongoing using the different types of gene therapy. Table 2
| Table 2: Studies of gene therapy in Sickle cell Disease5 | ||||||
| S/N | Type of gene therapy | Study name | Use of editing tools | Uses a vector | Genetic target | Protein product |
| 1 | Gene Editing | CLIMB | Yes: CRISPR-Cas9
RNP |
No | BCL11A related | HbF |
| PRECIZN-1 | Yes:
Zinc finger |
No | BCL11A
related |
HbF | ||
| 2 | Gene Addition | LentiGlobin | No | Yes | NA | HbAT87Q |
| DRERAGLOBE | No | Yes | NA | βAS3 (an anti-sickling β-globin protein) | ||
| MOMENTUM | No | Yes | NA | HbFG16D | ||
| 3 | Gene Silencing | Genetic silencing of BCL11a | Yes:
ShRNA |
Yes | BCL11A related | HbF |
| 4 | Gene correction | CEDAR | Yes:
HiFi CRISPR-Cas9 RNP |
Yes | Sickle mutation |
HbA |
The gene modification can be done, in-vivo (in the patient stem cell cell) or ex-vivo (outside the patient stem cells). Example of in-vivo modification is the gene editing seen in the Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) gene therapy.
In ex-vivo modification, the modified gene must be reintroduced into the patients haemopoietic stem cells. This requires a vehicle called the vector. The vector is usually a modified virus from which the infective part of the viral genetic material (nucleic acid) has been removed and replaced with the modified genetic material. Viruses are used because they can penetrate cell barriers and insert their genetic materials into host cells.
Genetic therapy removes the hurdle of looking for compatible donor as is required in haemopoietic stem cell transplantation. It also reduces the immunologic complications associated with HSCT. However, it does not remove the complications associated with chemotherapy seen in HSCT. These include infertility, mucositis, nauseas, loss of appetite and secondary tumours. In fact, the risk of secondary tumours may be high in gene therapy because of the possibility of transplanting genetically damaged HSC.5,9
In conclusion, Genetic therapy is promising, however, the real and potential risk involved is yet to be fully recognised. Secondly, though it offers a high potential for cure, cost may be a significant limiting factor to its uptake. For now, HSCT remains the most readily accessible cure for SCD.
References
- Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, et al. Gene Therapy in a Patient with Sickle Cell Disease. New England Journal of Medicine [Internet]. 2017 Mar 2 [cited 2022 Sep 22];376(9):848–55. Available from: https://www.nejm.org/doi/full/10.1056/NEJMoa1609677
- Rees DC, Williams TN, Gladwin MT. Seminar Sickle-cell disease. 2010 [cited 2022 Sep 21]; Available from: www.thelancet.com
- What is Sickle Cell Disease? | CDC [Internet]. [cited 2022 Sep 21]. Available from: https://www.cdc.gov/ncbddd/sicklecell/facts.html
- Gluckman E, Cappelli B, Scigliuolo GM, de la Fuente J, Corbacioglu S. Alternative donor hematopoietic stem cell transplantation for sickle cell disease in Europe. Hematol Oncol Stem Cell Ther. 2020 Dec 1;13(4):181–8.
- Kanter J, Falcon C. Gene therapy for sickle cell disease: where we are now? Hematology [Internet]. 2021 Dec 10 [cited 2022 Sep 22];2021(1):174–80. Available from: https://ashpublications.org/hematology/article/2021/1/174/482932/Gene-therapy-for-sickle-cell-disease-where-we-are
- Olowoyeye A, Okwundu CI. Gene therapy for sickle cell disease. Cochrane Database of Systematic Reviews. 2020 Nov 30;2020(11).
- Orkin SH, Bauer DE. Emerging Genetic Therapy for Sickle Cell Disease. https://doi.org/101146/annurev-med-041817-125507 [Internet]. 2019 Jan 28 [cited 2022 Sep 22];70:257–71. Available from: https://www.annualreviews.org/doi/abs/10.1146/annurev-med-041817-125507
- Demirci S, Uchida N, Tisdale JF. Gene Therapy for Sickle Cell Disease: An Update. Cytotherapy [Internet]. 2018 Jul 1 [cited 2022 Sep 22];20(7):899. Available from: /pmc/articles/PMC6123269/
- Hoban MD, Orkin SH, Bauer DE. Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood [Internet]. 2016 Feb 18 [cited 2022 Sep 22];127(7):839–48. Available from: https://ashpublications.org/blood/article/127/7/839/35247/Genetic-treatment-of-a-molecular-disorder-gene
- Lettre G and Bauer DE. Fetal haemoglobin in sickle-cell disease: from genetic epidemiology to new therapeutic strategies. Lancet. 2016. 387(10037), 2554-2564. DOI: 10.1016/S0140-6736(15)01341-0


{kind=link}
{kind=link}
{kind=link}