
SICKLE CELL DISORDER: IMPORTANCE OF SELF-CARE (PART ONE)

The Importance of Nutrition in the Management of SCD
June 16, 2023
PIPSICKLE TRIAL: The journey so far
June 16, 2023
SICKLE CELL DISORDER: IMPORTANCE OF SELF-CARE (PART ONE)
Prof. Norah O. Akinola, FACP
PhD, FMCP, FMCPath
Department of Haematology and Blood Transfusion,
Obafemi Awolowo University Teaching Hospitals Complex,
Ile-Ife, Osun State, Nigeria.
Our honourable Chairman of the today’s occasion, Dr. Femi Mobolaji-Lawal, our wonderful Chairman of the Board, Prof. Olu Akinyanju, a mentor of mine and one of the haematologists who received me when I returned to Nigeria in 1992. He is a wonderful teacher, Professor of Professors, I doff my cap to you Sir. Congratulations for today. I also want to thank your darling wife who I also knew very well, thank you very much for what you are doing for him. You are looking much younger than I expected, so he must be doing something for you too.
Some of you had not come in when the orchestra was playing. They were absolutely wonderful. Give them a round of applause please. You will go places, thank you very much. It reminded me of the time I visited the NIH, National Institutes of Health, Bethesda, Maryland, USA, in my quest to acquire knowledge in bone marrow transplantation. In the Transplant Building, there, in the reception, after the secretary and other administrative staff, is the place where the orchestra plays. They play morning, afternoon and evening, it is simply awesome. The performance of the orchestra this morning reminded me of that time when I was in NIH, please give them another round of applause.
So, I have been asked to give a talk about, “Sickle Cell Disorder: Importance of Self-Care”. This year June 19 was shared by something special; it was shared by Father’s Day. It’s not a coincidence and I am sure it was for a purpose. I would like to recognise all fathers here today and those who are online, at home and in Diaspora, especially our grandfather, Prof. Olu Akinyanju, our chairman of the day and our honourable gentleman from the WHO, thank you very much, you are all fathers, we appreciate you. But there is a father, I should say, “was”, now, because he is now late, who was a father of research in sickle cell disease (SCD), who I would like to pay a tribute to this morning, that is our very own Prof. Kwaku Ohene-Frempong, from Ghana. He spent most of his working years at the Children’s Hospital, Philadelphia, USA, but he refused to become an American citizen. He did not change his citizenship and was a Ghanaian to the core until he died. He contributed immensely to sickle cell research, internationally. He single-handedly started newborn screening in Ghana, in 1993 and recently he made a video to increase awareness for SCD where he stated categorically that, “SCD is manageable, it is not a death sentence”. So, if you don’t hear anything that I say today, please go home with that.
Sickle cell disease is manageable, it is not a death sentence, that is a tribute to Prof. Ohene-Frempong of blessed memory. May his soul rest in peace. At that point I will ask you to do a little bit of exercise and stand for one minute of silence. For all our departed, whether they were healthcare providers or caregivers at homes of people living with SCD, one minute silence please. May their gentle souls rest in peace. Amin.
Everybody has been hearing about SCD and you will be hearing about it over, and over, and over again, and it is for emphasis. Sickle cell disease is a global public health concern and Nigeria has the largest population of people living with SCD in the world or being born with SCD (150 babies affected) annually. It is a non-communicable disease (NCD) and therefore it should be of great concern to our government at all levels. We have been celebrating the World sickle cell day, June 19, since 2010, when it was to commemorate 100 years of the paper that was written to first justify the use of the word, “sickle cell”, because previous papers had described features seen in people in various places, but that was the first paper to describe sickle cell as we see it under the microscope and as we know it today. That paper was written by James Herrick and his team in November 1910, entitled, “Peculiar Elongated and Sickle-shaped Red Blood Corpuscles in a Case of Severe Anemia”. It all started in December 1904 when Walter Clement Noel a 20 year old man from Grenada went to the USA to study dentistry at the Chicago College of Dental Surgery. He first sought medical attention at the Presbyterian Hospital and was treated by Dr. Ernest E. Irons, an intern training under Dr. James Herrick. Dr. Irons took his blood and had a look at it and saw these strange cells, which are now known as sickle cells. As usual it is not the intern that gets the glory, but it is the oga (boss) at the top. This slide (Figure 1) shows irregular cells similar to those seen under the microscope, so it was Herrick et al, 1910 that got the credit. Since then so many papers have been published on SCD.
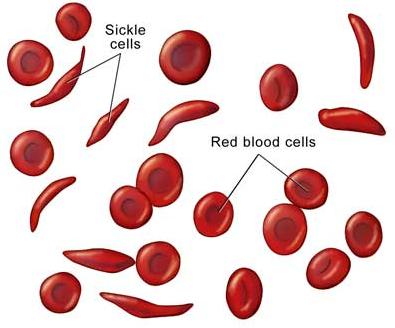
Figure 1. Sickle cells seen in the peripheral blood
Figure 2 is a schematic representation of SCD of the prevalence of sickle cell disorder in Nigeria, as you can see here, about 25% are carriers and I am one of them, that is I am one of the one in four that carry the sickle cell gene and I am not stigmatised because I stigmatise myself first, by saying, at the time I had to choose a life partner, “everybody, I am a carrier, if you are a carrier don’t come near me, so if you are not AA don’t apply”. So that was it and there was a reason for it, because the last born of my mother had the disease and she died at the age of 12 years. She had it really bad and we were living in London at the time and at the age of six months when she was to begin to show the signs and symptoms of the disease, she got the most severe presentation of all, meningococcal meningitis. The Paediatricians will understand what I mean by that, but she survived it because we were in an environment where they knew what to do for her. She was immediately put on penicillin prophylaxis, folic acid and the lot, but that does not change the severity of the disease that she was born with. She had inherited a severe disease and that infection she had should have caused her to either be deaf or have other neurological deficits, but because she was managed properly, she came out of it completely whole without any complications. That is why I have spent 30 years of my life if not more studying SCD in various ways.
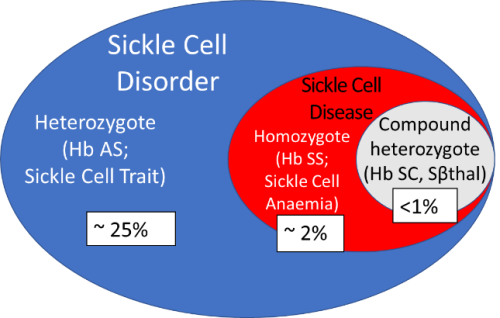
Figure 2. Schematic representation of sickle cell disorder (not to scale) showing the prevalence in Nigeria.
DEFINITION OF TERMS AND MYTHS
I will start by giving definition of terms to make it easy for those who are not medics amongst us to follow the talk. I can see many of my colleagues here who know everything about SCD, so this talk is not for you, it is for those who need information, who need the education.
- Haemoglobinopathy is an inherited disease associated with qualitative and /or quantitative abnormality in the production of haemoglobin, the protein that delivers oxygen to the tissue and takes carbon dioxide from the tissue to the lungs for excretion.
- Sickle cell disease is a qualitative abnormality of haemoglobin, whilst thalassaemias are quantitative abnormalities.
- Sickle cell disorder is the inheritance of at least one single sickle haemoglobin gene, it therefore includes Hb AS (heterozygote; sickle cell trait; SCT).
Sickle cell trait (Hb AS), is a carrier state and not a disease. It is usually not associated with symptoms of crises, however, under stressful conditions or low oxygen tension (hypoxia), a carrier may have mild pain or some traces of blood in urine (microscopic haematuria).
Haemoglobin S Haplotypes
Haemoglobin S haplotypes are the genes that are responsible for making the haemoglobin molecule, that is, the genes that produce the alpha, beta, delta and gamma globin chains.
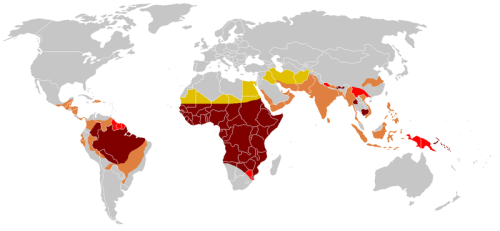
The Hb S haplotype is variable and may be
- Benin (BEN): associated with the lowest %Hb F; it has the most severe disease profile and occurs in 97% of Nigerians;
- Senegal (SEN);
- Bantu (Central African Republic, CAR); has intermediate %Hb F
- Cameroon (CAM); or
- Arab-Asia (ARAB): has high %Hb F that is associated with a mild disease profile.
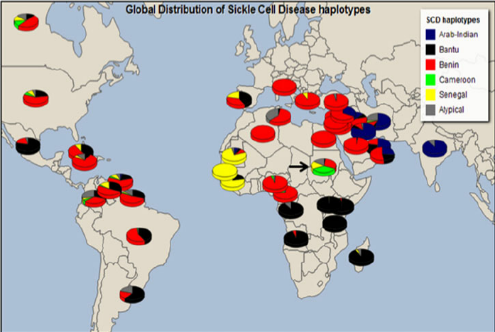
Valentina J. et al, (2015) A Journal of Integrative Biology VOL. 19, NO. 3 | https://doi.org/10.1089/omi.2014.0134
Figure 3. Global distribution of SCD haplotypes.
Genetic finger printing is DNA analysis that confirms the diagnosis of SCD. When used to determine the diagnosis of an unborn child at around 10 weeks gestation, it is called prenatal diagnosis (PND).
Table 1. Some Usual and Unusual Haemoglobin Types
| Phenotype | %Hb A | %Hb S | %Hb F | %Hb A2 |
| Usual Feotus (Hb F) | 5-10 | – | 85 | – |
| Usual Adult (Hb A) | 97 | – | <1 | 2-5 |
| Trait (Hb AS) | 60-75 | 25-40 | <1 | ~3 |
| SCA (Hb SS) | 0 | 75-90 | 5-20 |
- Hb A (a2b2) – is the usual (major) adult Hb
- Hb F (a2g2) – is the usual (major) foetal and a minor adult Hb
- Hb A2 (a2d2) – is a minor adult Hb, which increases in beta-thalassaemia
Hb S – is an unusual, but major adult haemoglobin occurring as a result of a single point mutation at the 6th position of the b globin gene in which glutamic acid (hydrophilic; GAG) has been changed to valine (hydrophobic; GTG)
Hb C – is an unusual adult haemoglobin also occurring as a result of a mutation at the 6th position of the b globin gene – lysine (AAG) instead of glutamic acid
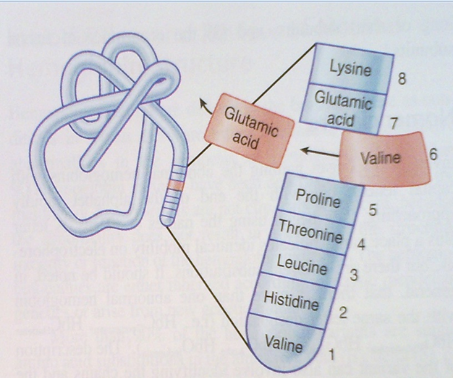
Figure 4. Genetic mutation in Hb S involving the substitution of valine for glutamic acid at position 6 of the beta globin gene.
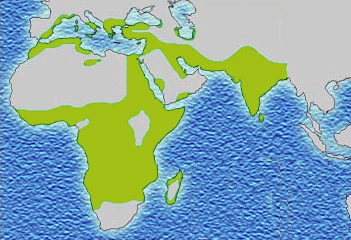 Modern distribution of malaria |
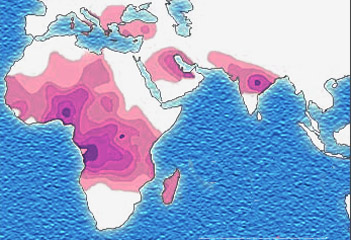 Distribution of the sickle cell trait shown in pink and purple |
 Historical distribution of malaria (no longer endemic in Europe) shown in green |
|
Figure 5. Endemicity of Malaria and Sickle Cell Trait (SCT) worldwide.
Figure 5 shows that the areas of the world associated with endemic malaria are also the areas where the sickle gene predominates, i.e. mainly in the tropics, Sub-Saharan Africa and some parts of Asia.
Effect of Malaria Infection on Red Blood Cells
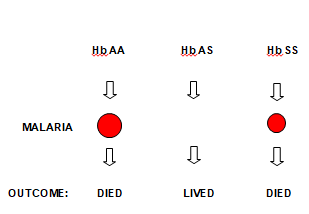
Figure 6. The Concept of Balance Polymorphism
Before the discovery of chloroquine for the treatment of malaria, people with haemoglobin AA used to die of severe anaemia following malaria infection. The persistence of malaria caused the genetic mutation that results in the production of sickle haemoglobin, which prevents malaria parasite from growing in and destroying red blood cells. This genetic mutation is transferred from one generation to another and individuals inheriting Hb AS are protected from malaria and its complications. This is the concept of balance polymorphism that results in survival advantage of individuals with Hb AS (Figure 6).
Myths
There are many myths and beliefs about SCD. Some of them include the following:
- Affected individuals do not live beyond the age of 21 years, “abiku”.
- They are spiritually possessed, “ogbanje”.
- They are infertile.
- They cannot do any form of exercise.
ALL THESE ARE FALSE BELIEFS AND ARE NOT TRUE!!!
Pattern of Inheritance
Table 2. Putten Squares: A Game of Chance
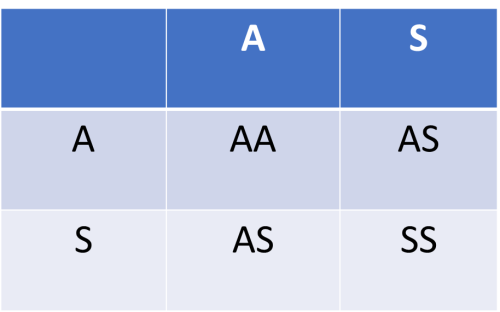 (1) 25% chance for every pregnancy to be Hb AA or Hb SS, but a 50% chance of being Hb AS |
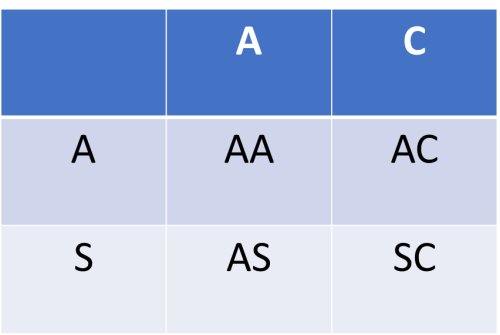 (2) 25% chance for every pregnancy to be Hb AA or Hb SC or Hb AS or Hb AC |
The inheritance of the sickle cell gene is by Mendelian fashion. This means that each gene from a parent has an equal chance of being inherited by the offspring. If both parents are carriers of the sickle gene (i.e. Hb AS) as in Table 2 (1) the child has 50% chance of having Hb AS; and 25% chance of having Hb AA or Hb SS for each pregnancy. If one parent is a carrier of the sickle gene (Hb AS) and the other is a carrier for haemoglobin C gene (Hb AC), there is 25% chance that the child may have Hb AA, Hb AS, Hb AC or Hb SC for each pregnancy. So, the inheritance of any unusual haemoglobin (variant) is a game of chance. Since prevention is better than cure, everyone should know his/her haemoglobin type as this would enable informed decision making when it comes to having babies. Do not play the chance game with your babies.
MORE DEFINITIONS
What is the Steady State?
- Steady State: a pain free period of at least 3 weeks after an event of acute pain and at least one week before the onset of another acute episode; at least three months after an intervention like blood transfusions or use of disease modifiers.
- The steady state is not steady rheologically, because there are inter- and intra-personal variations that simulate crisis without overt pain, but which cause changes in clinical presentations with time, depending on the severity of the disease.
What are Crises?
- Crises are phases of SCD that punctuate the steady state.
- Painful crisis – most common, also called vaso-occlusive crisis (VOC).
- Haemolytic crisis – increased destruction of red cells that worsens the degree of anaemia and deepens jaundice.
- Sequestration crisis – pooling of blood in the spleen or liver and sudden worsening of anaemia. (Mothers should always examine the abdomen of their babies to check the size of the spleen.)
- Aplastic crisis – usually due to infection by Parvovirus B19 that destroys red cell precursors and worsens the anaemia.
Megaloblastic crisis – deficiency in folic acid and/or vitamin B12.
To be continued in Part 2


{kind=link}
{kind=link}
{kind=link}